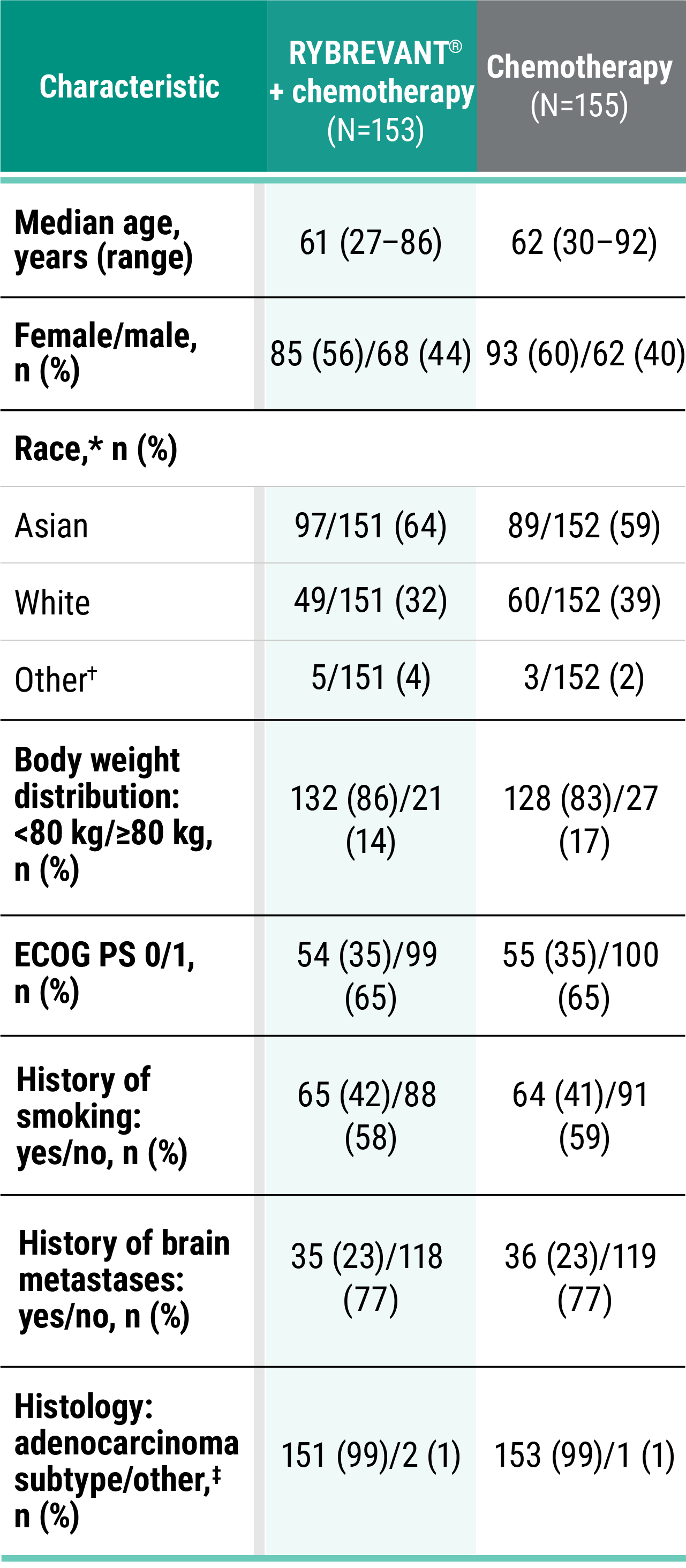
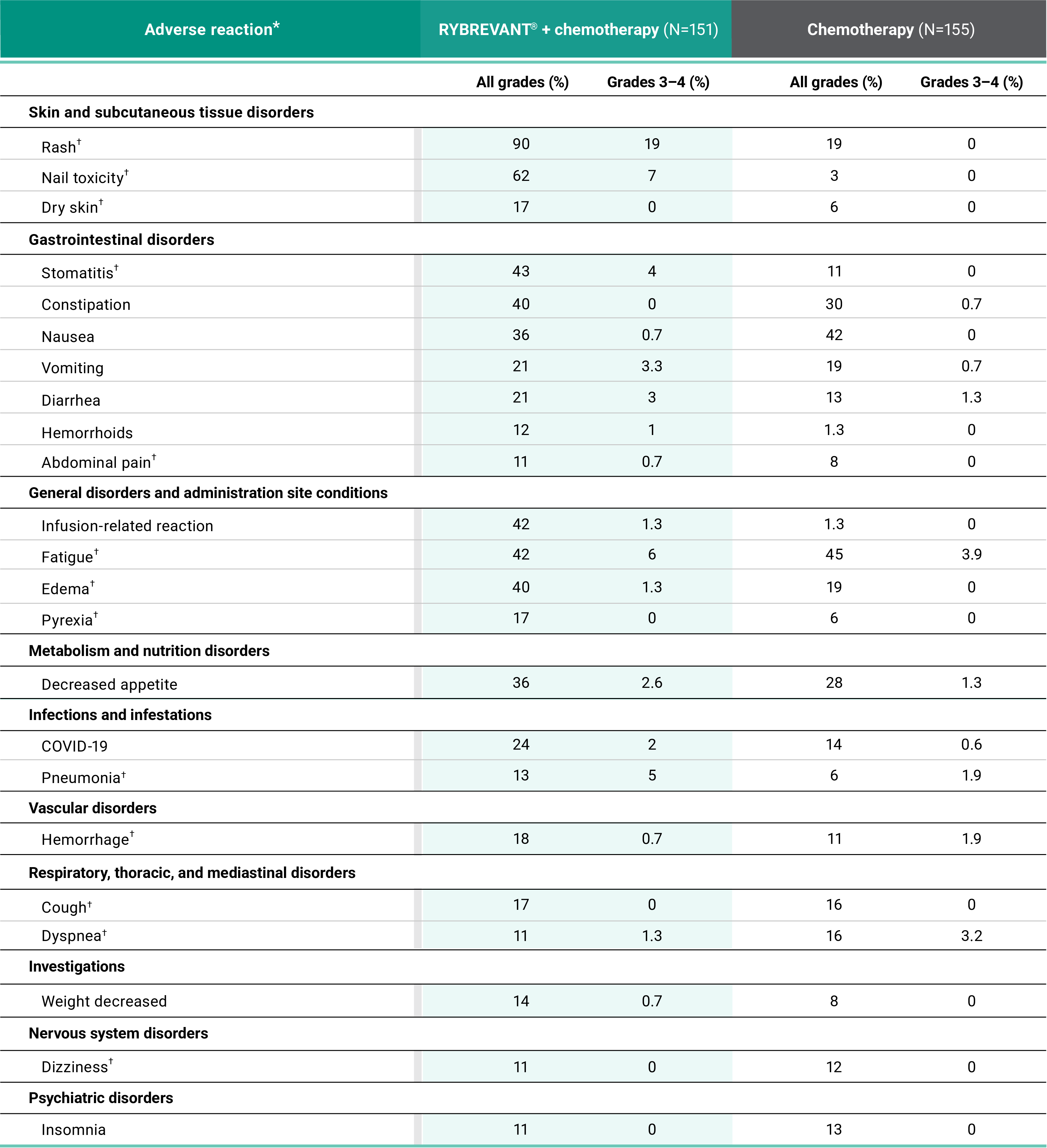
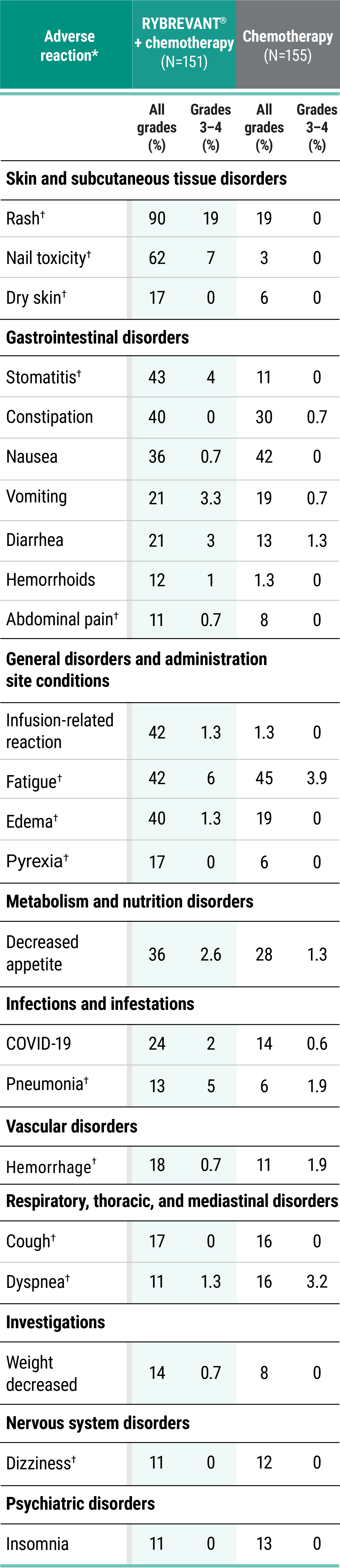
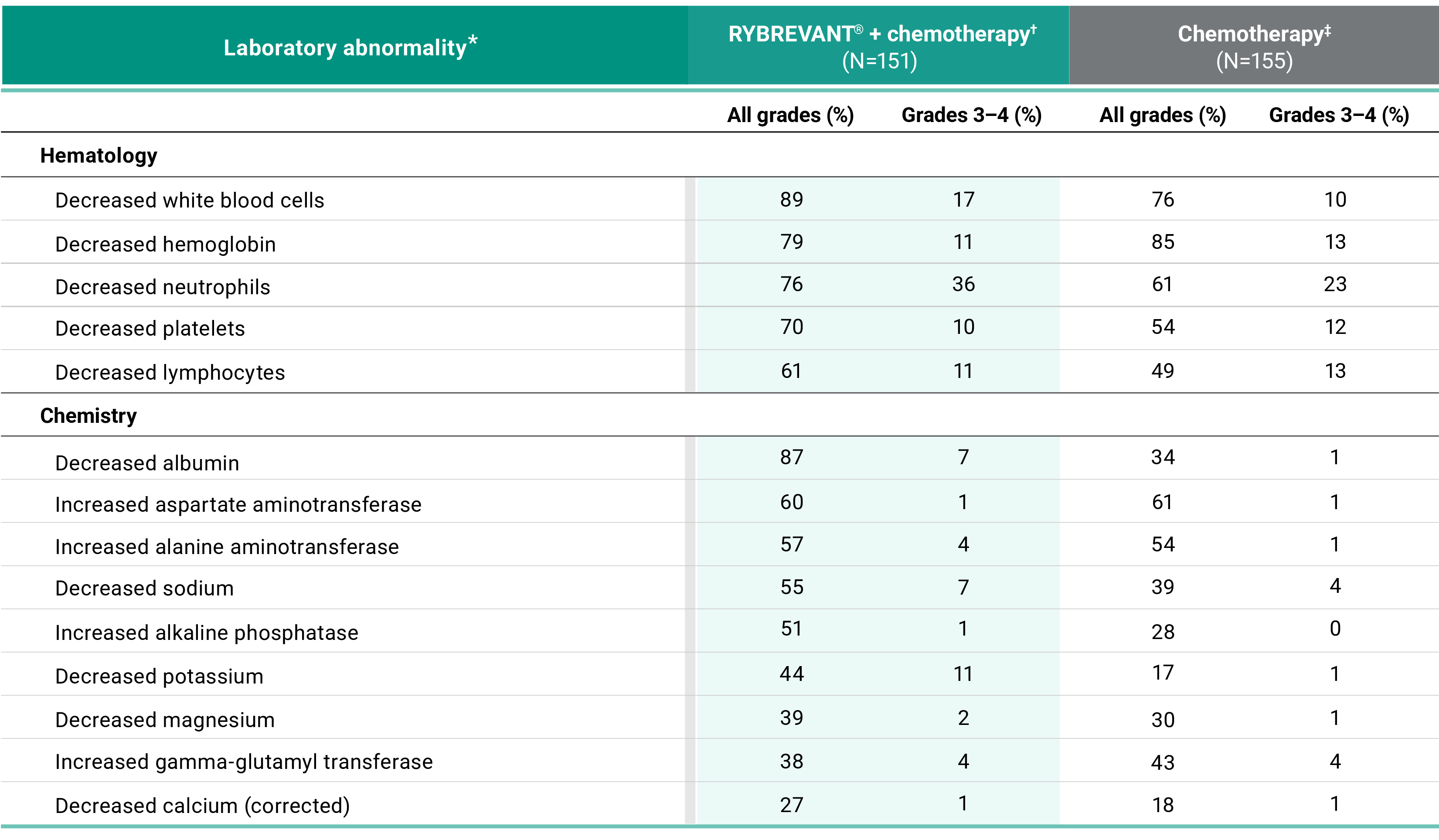
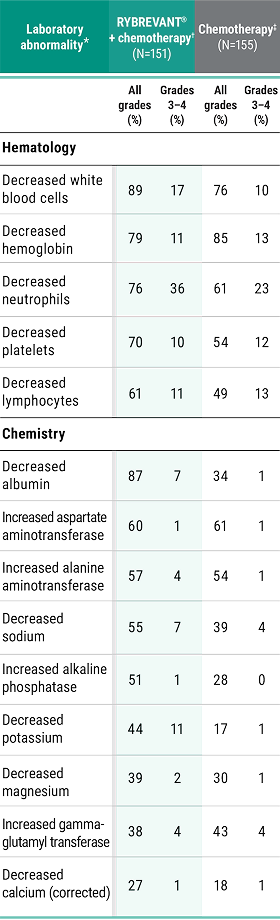
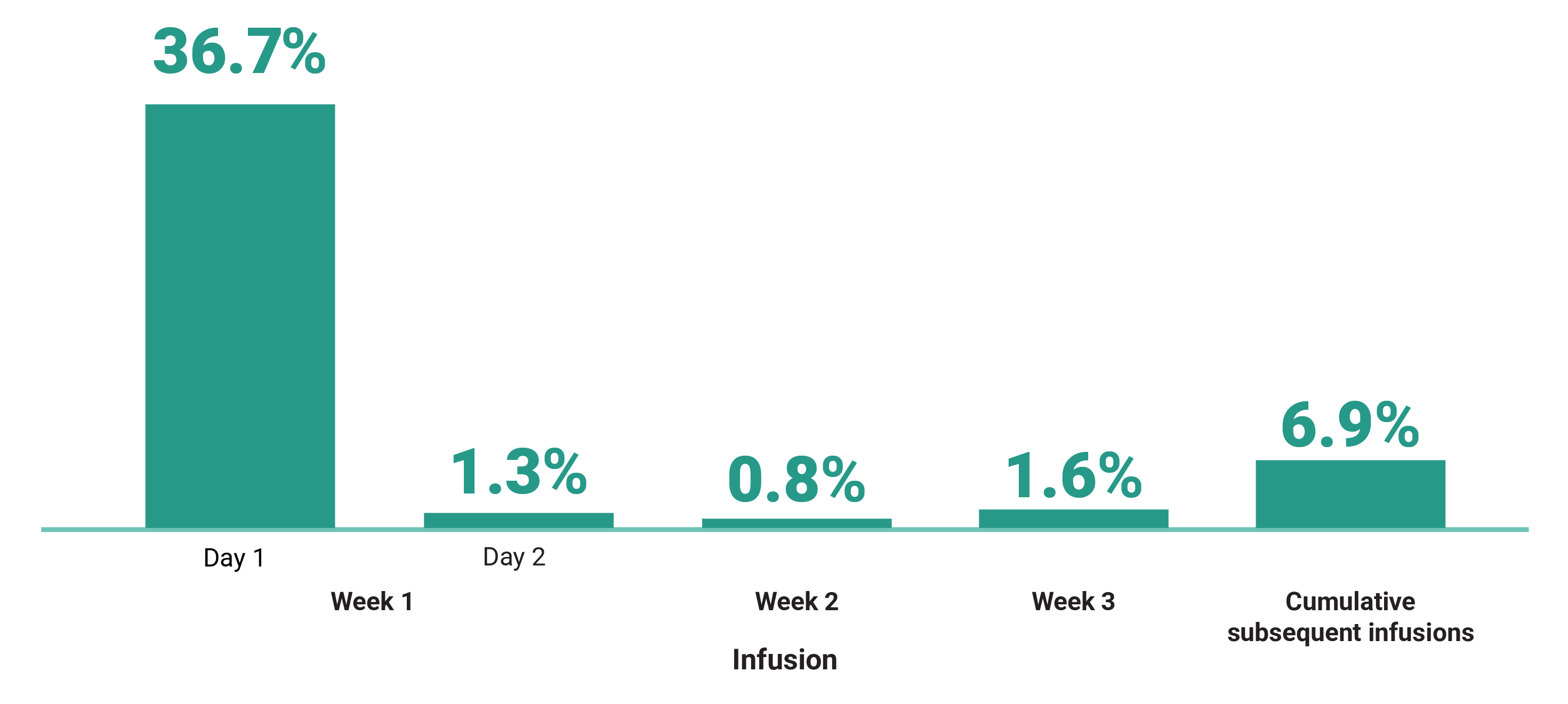
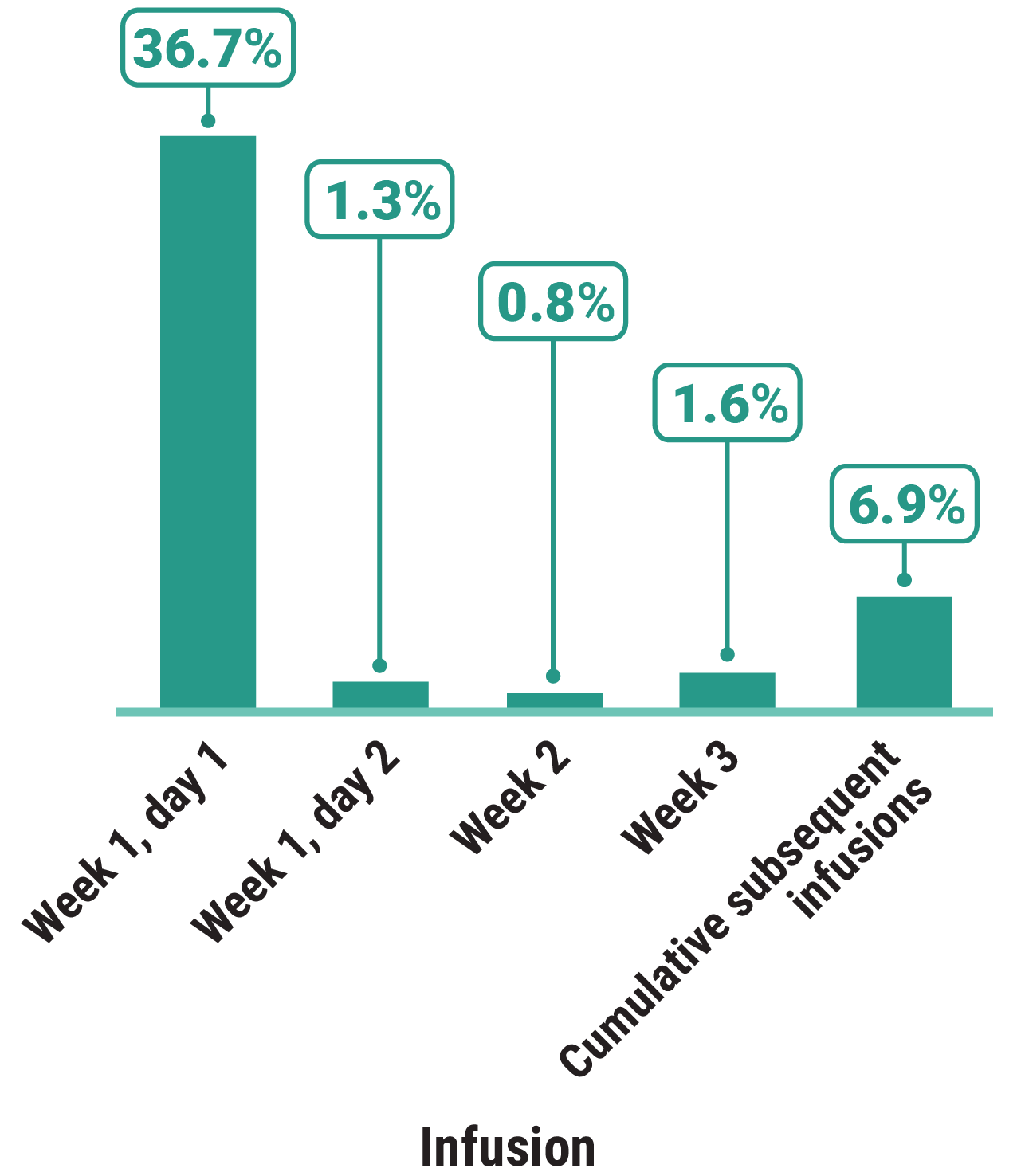
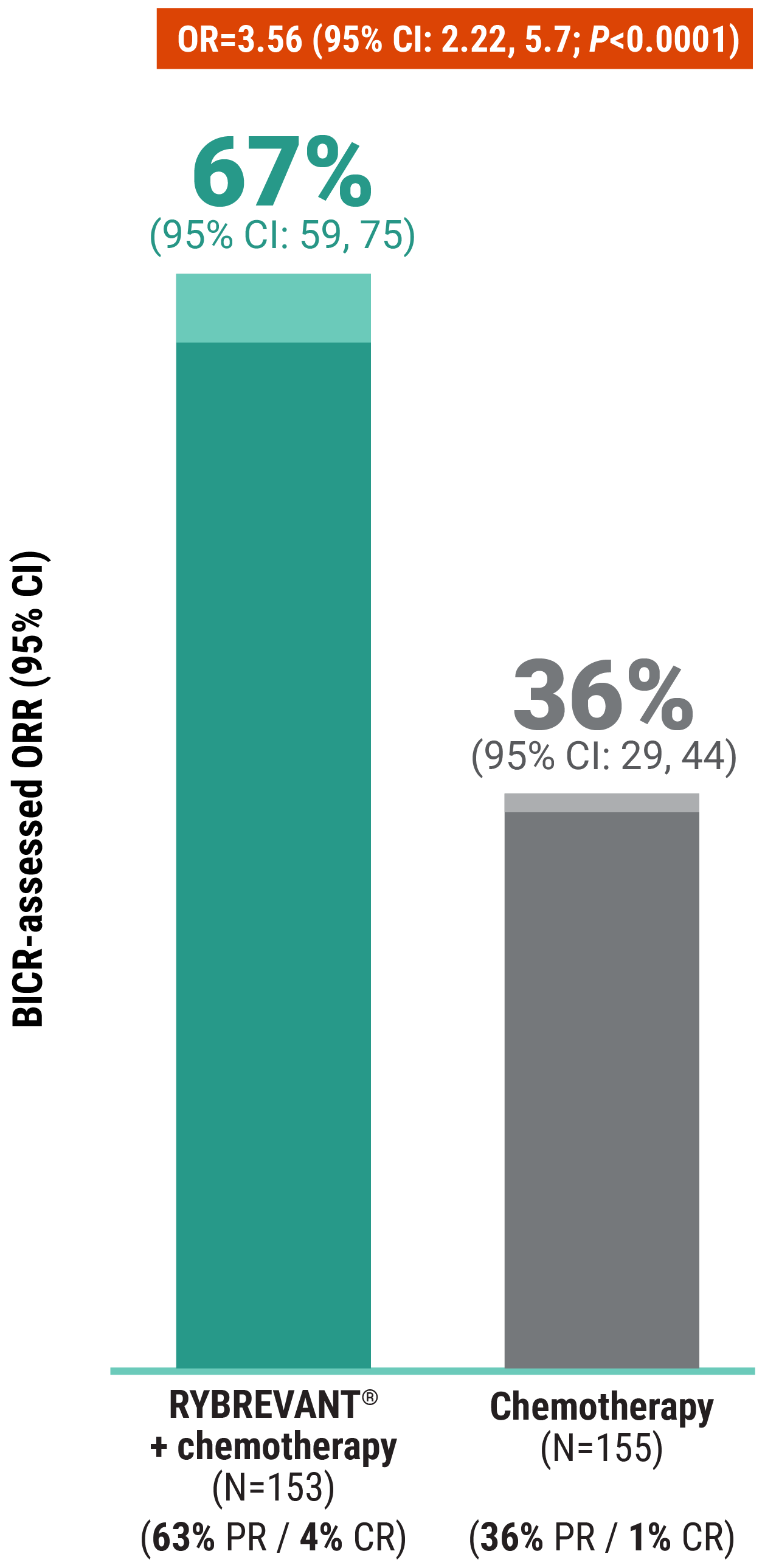Primary Endpoint
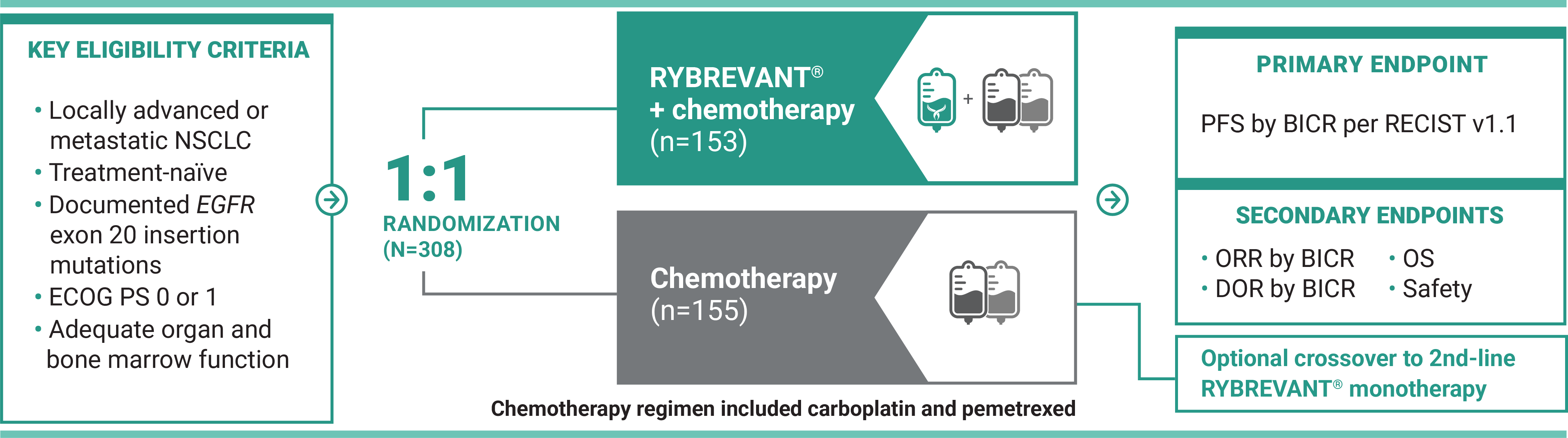
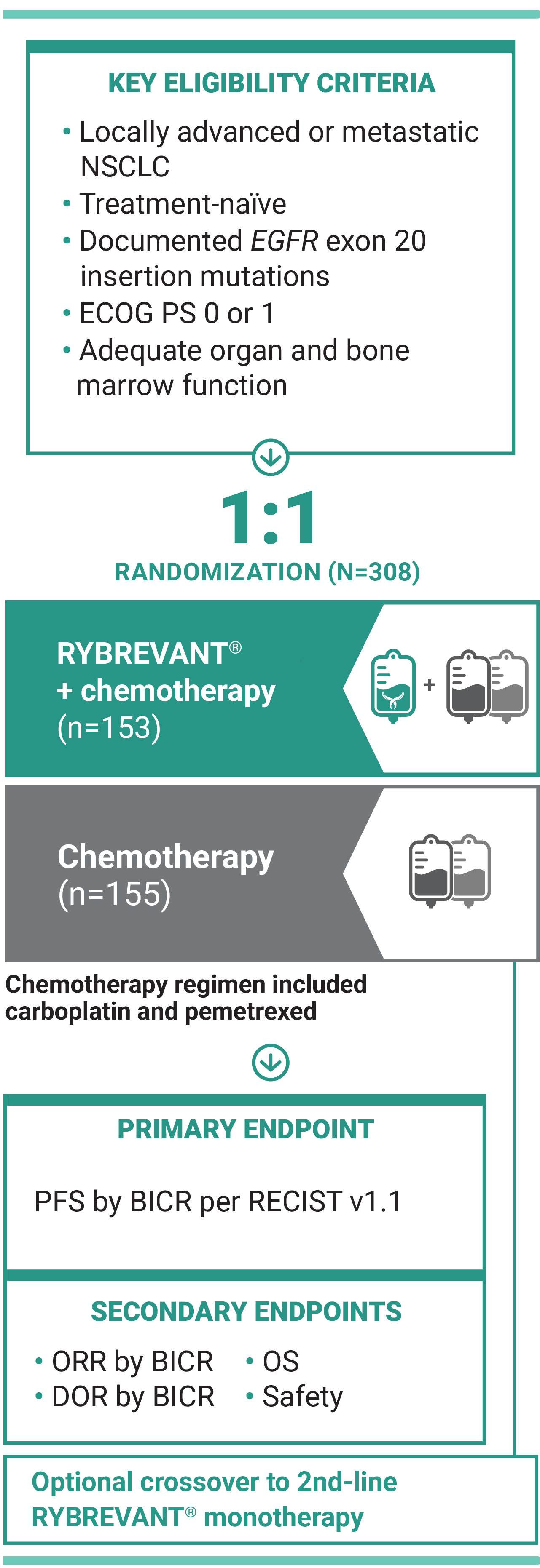
For first-line treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations
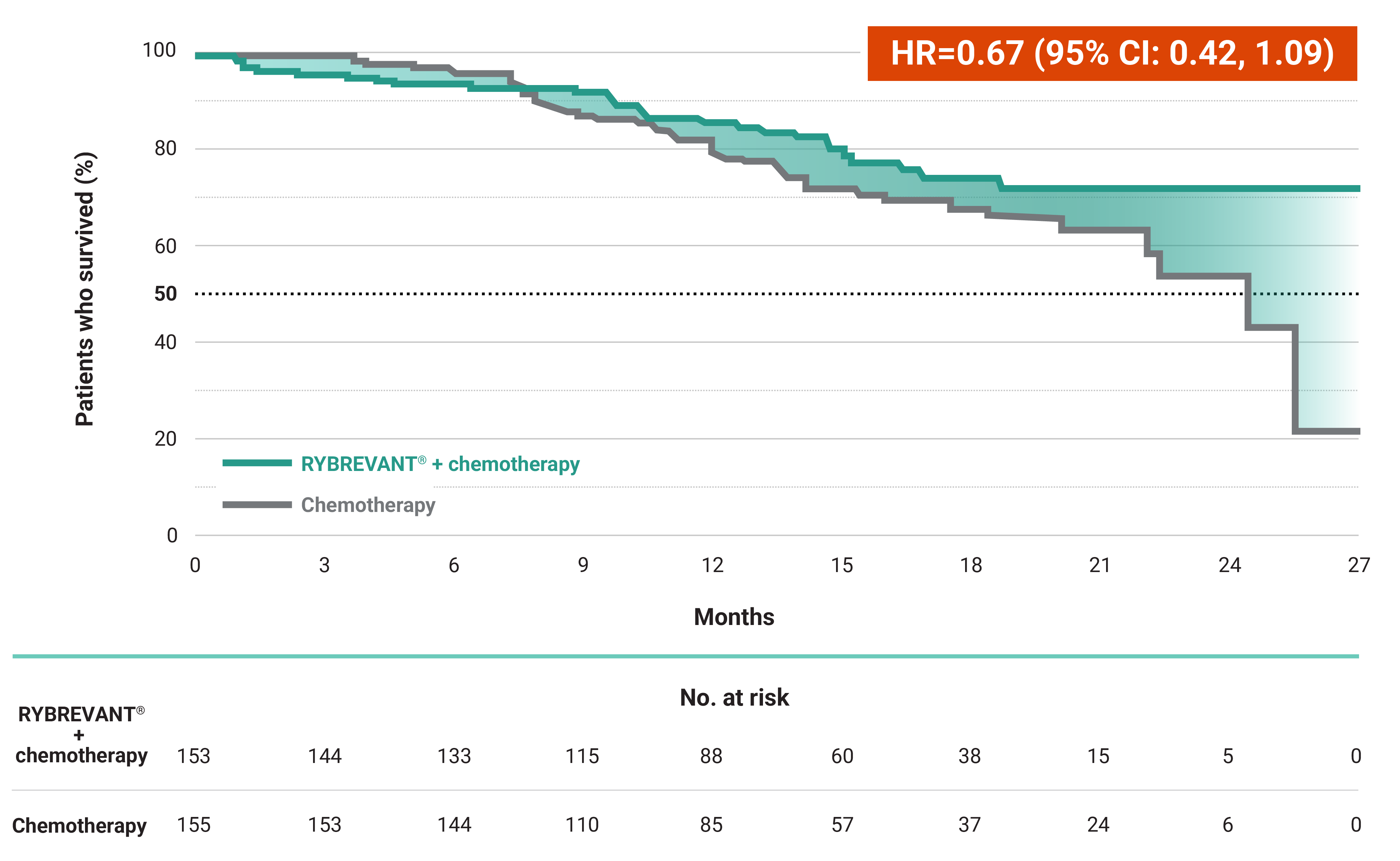
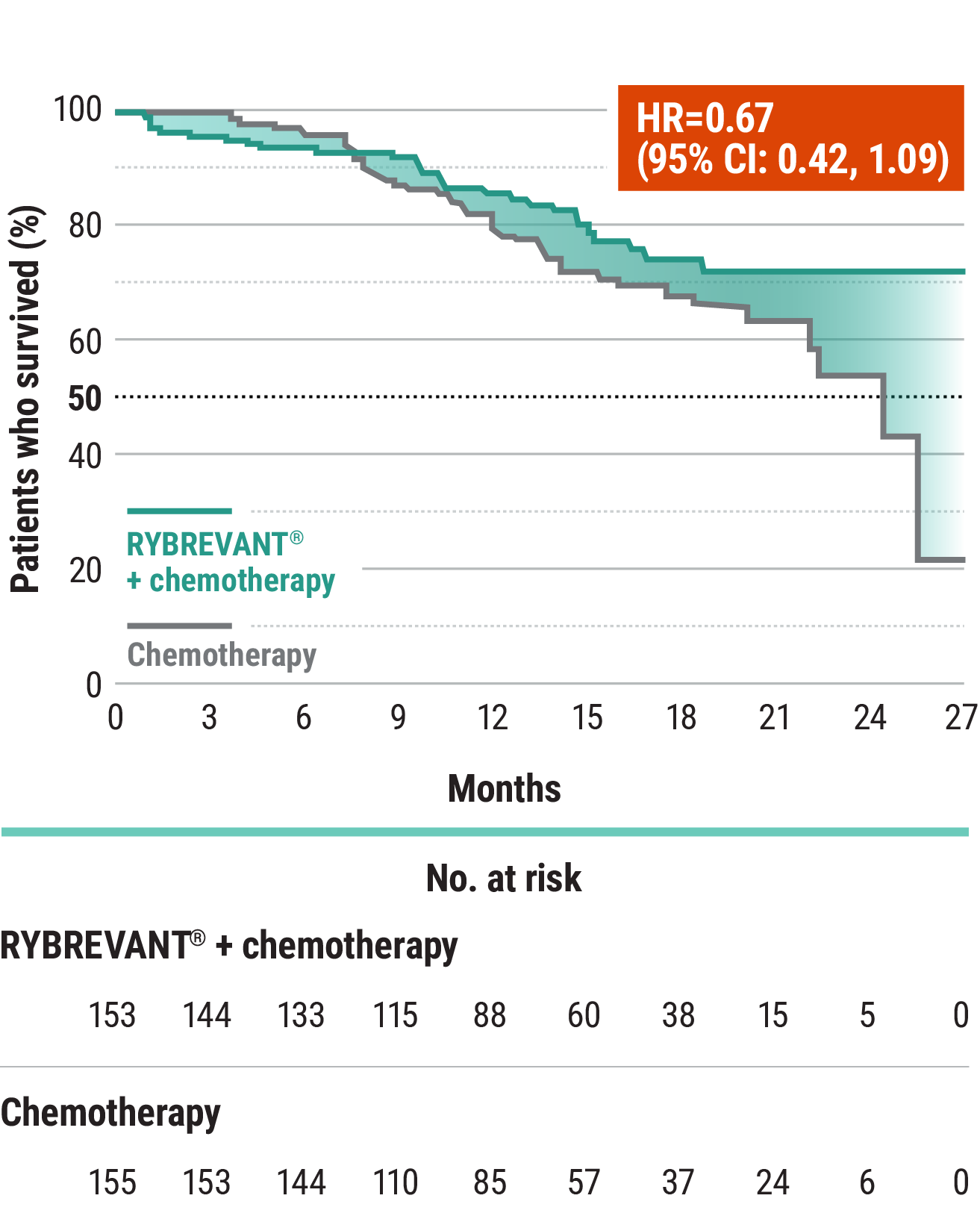
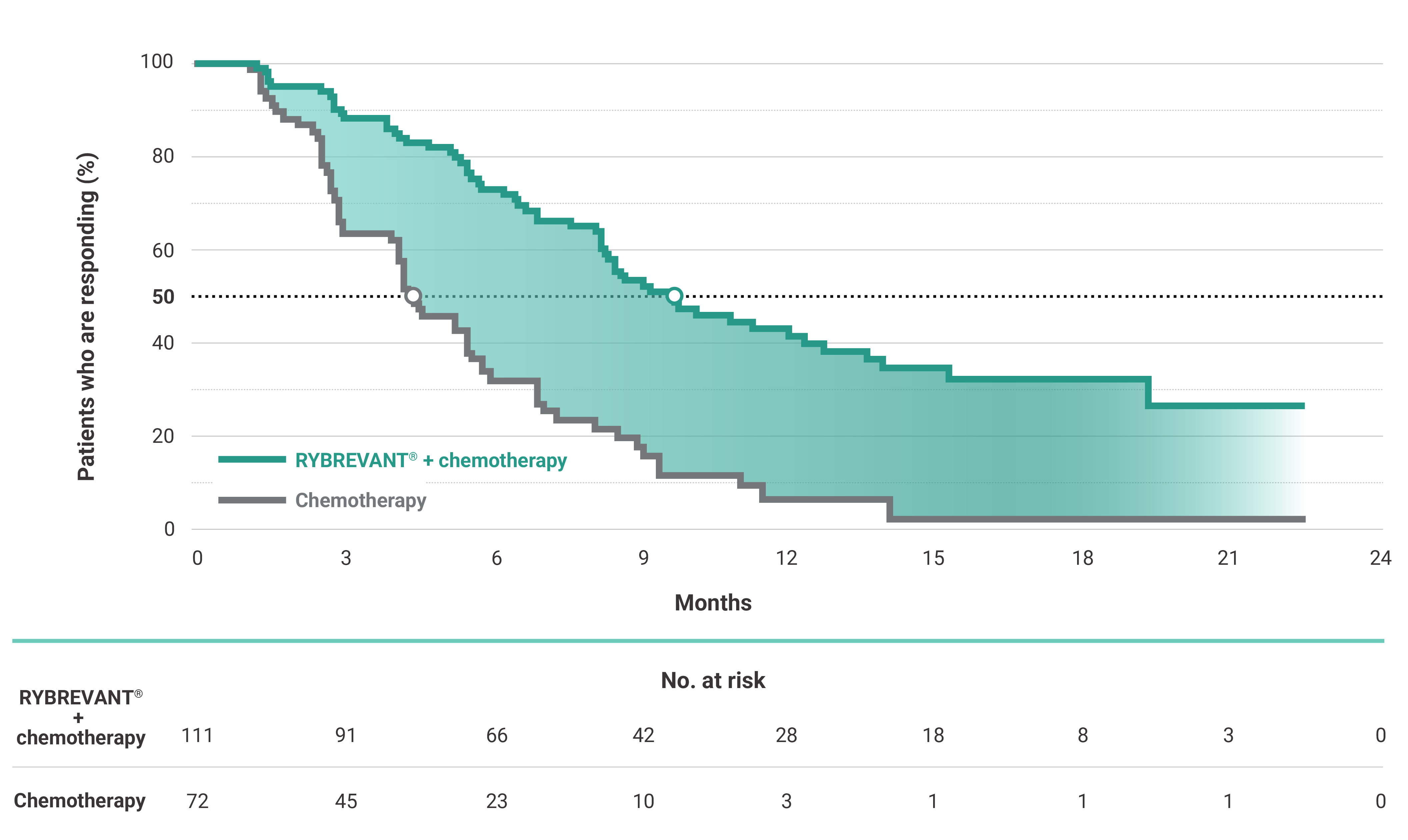
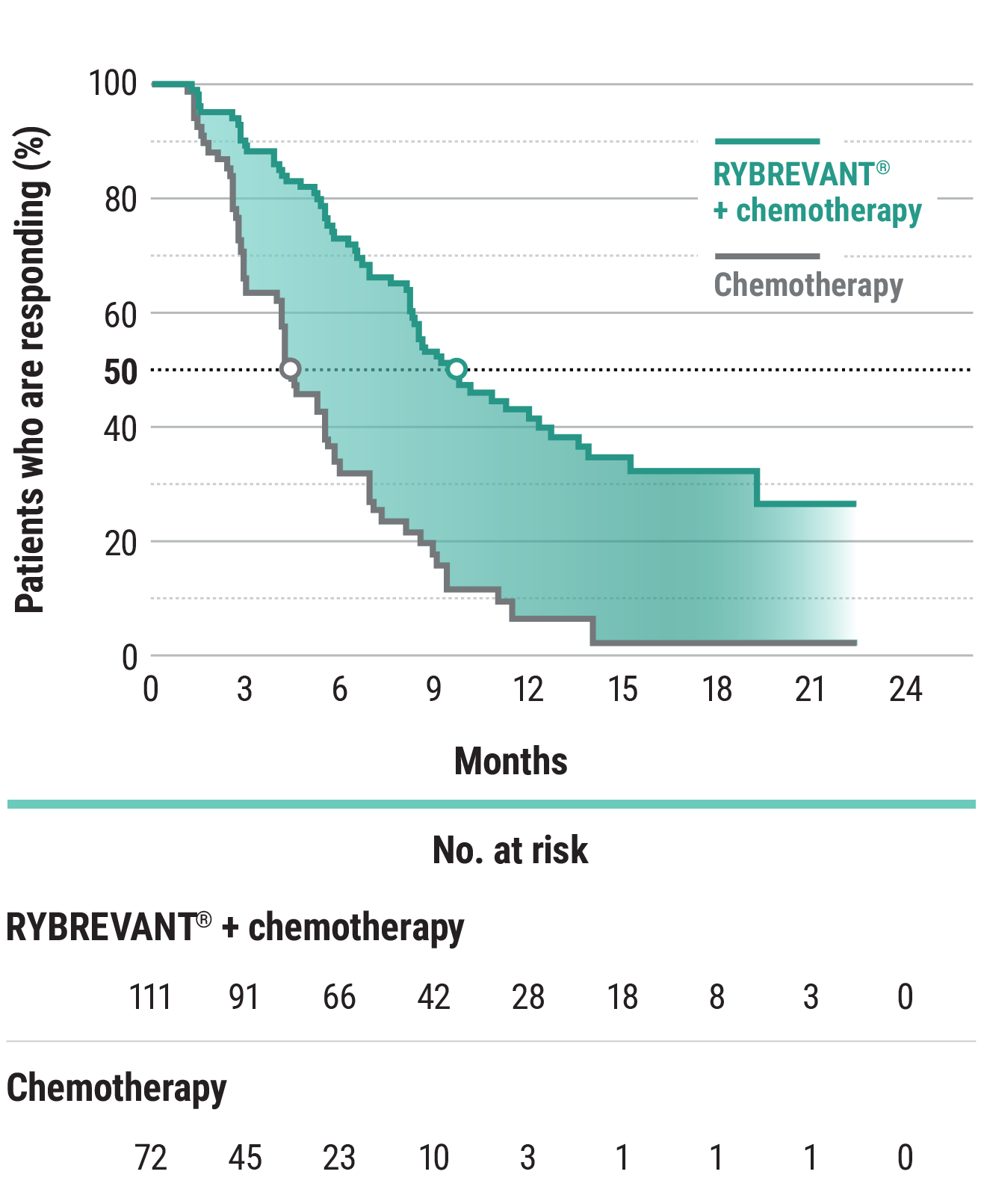
The first and only targeted therapy to show statistically significant PFS improvements1
RYBREVANT® + chemotherapy demonstrated a statistically significant reduction in the risk of progression or death by 60% vs chemotherapy alone, as assessed by BICR1,2
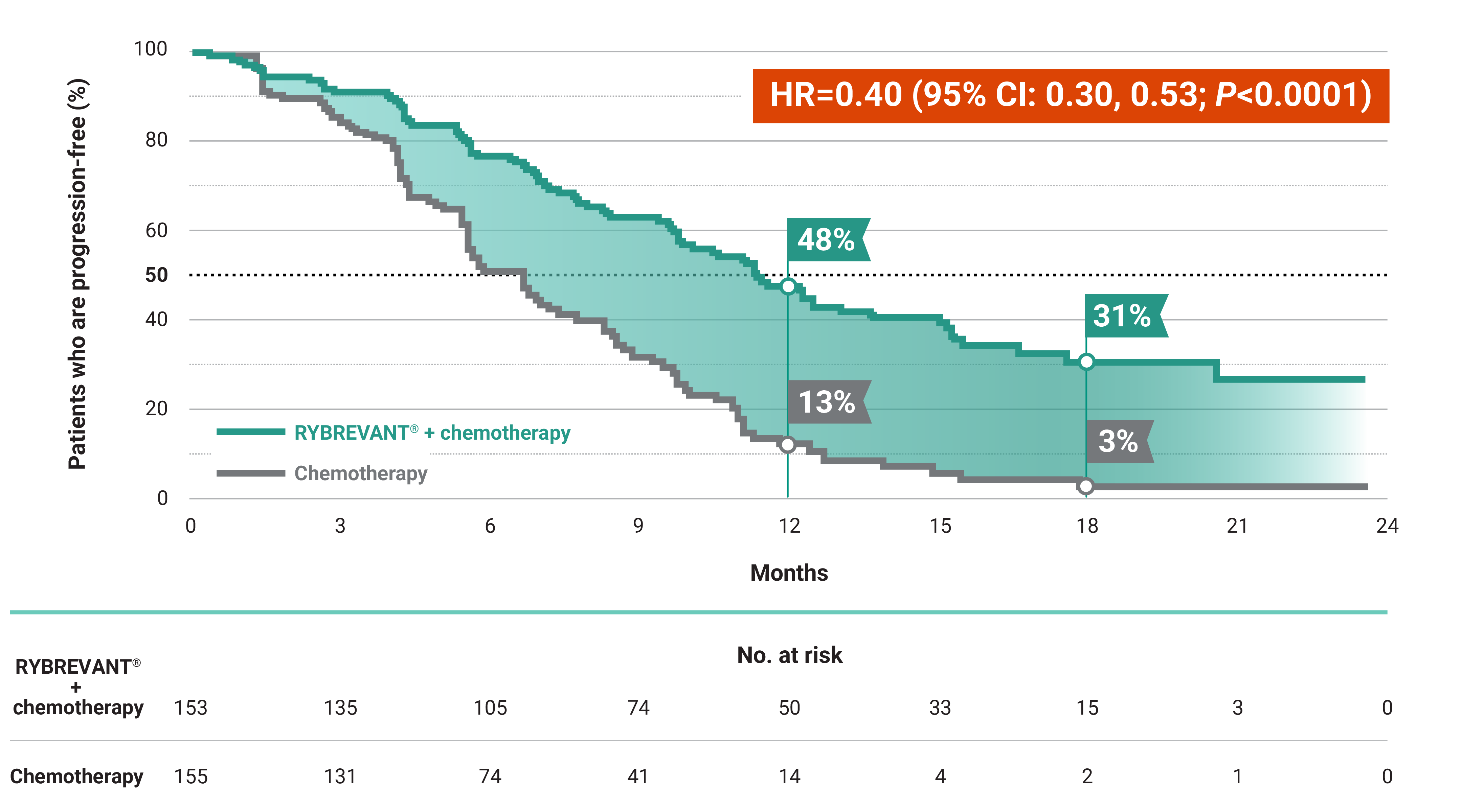
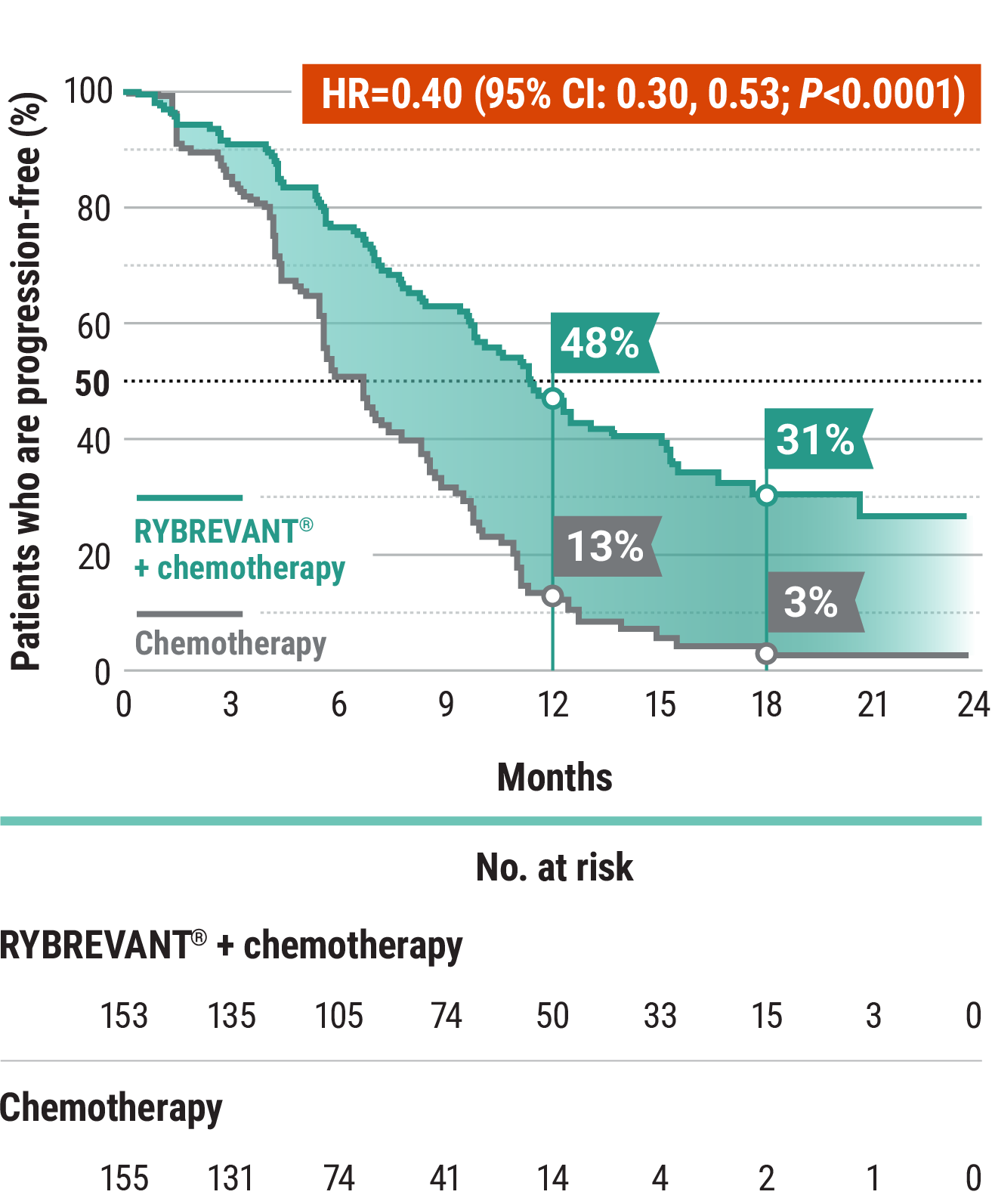
~2X mPFS: 11.4 months with RYBREVANT® + chemotherapy vs 6.7 months with chemotherapy alone1
*Amivantamab includes both amivantamab and hyaluronidase-lpuj (RYBREVANT FASPRO™) subcutaneous injection and IV amivantamb-vmjw (RYBREVANT®). Amivantamab and hyaluronidase-lpuj has different dosing and administration instructions compared with IV amivantamab-vmjw.3
†Chemotherapy is carboplatin and pemetrexed.3
PFS results in prespecified subgroups2


This was a prespecified analysis and was not powered to show statistical significance.
RYBREVANT® is also approved for patients with EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy1
Learn moreBICR, blinded independent central review; CI, confidence interval; ECOG PS, Eastern Cooperative Oncology Group performance status; EGFR, epidermal growth factor receptor; HR, hazard ratio; IV, intravenous; mNSCLC, metastatic non–small cell lung cancer; mPFS, median progression-free survival; NSCLC, non–small cell lung cancer; PFS, progression-free survival.